

Różnica między zespołem Turnera a zespołem Downa

- 2612
- 771
- Marta Ruciński
Zespoły Down i Turnera są zarówno nieprawidłowościami chromosomowymi, ale różnią się pod względem chromosomów, a także ich rozpowszechnienie, przyczyny, cechy, diagnoza i rokowanie.
Rozpowszechnienie
Zespół Downa (DS), po raz pierwszy opisany przez Johna Langdona Down, angielskiego lekarza pod koniec XIX wieku, jest wynikiem pełnej lub częściowej kopii Chromosomu 21, z którą rodzą się 1 na 700 dzieci w Stanach Zjednoczonych (zespół krajowy Down Downa Społeczeństwo). W przeciwieństwie do tego, zespół Turnera (TS) występuje wyłącznie u kobiet z jednym zamiast dwóch chromosomów X. Około 1–2% zarodków ma TS, ale 99% tych zarodków będzie spontanicznie przerywać, najczęściej w pierwszym trymestrze trymestru, co prowadzi do około 1 na 2500 urodzeń żywych na całym świecie (Departament Zdrowia i Opieki Społecznej USA).
Powoduje
Przyczyna dodatkowego częściowego lub pełnego chromosomu w DS pozostaje nieznana, ale wiek matki został zidentyfikowany jako czynnik. Na przykład 20-letnia matka ma 0.1% szansa na poród dla niemowlęcia z DS, podczas gdy 45-letnia matka ma 3% szansę (j. K. Morris i in.). W około 5% przypadków pochodzenie może być powiązane z ojcem.
Zidentyfikowano kilka rodzajów DS.
- Trisomia 21 występuje, gdy przed lub podczas poczęcia para chromosomów 21 w nasieniu lub jajdzie nie oddziela się, co powoduje trzy kopie chromosomu 21, a nie dwa. One powtórzają się w każdej komórce ciała.
- Translokacja DS jest widoczna w około 4% przypadków. Tutaj liczba chromosomów wynosi 46, ale pełna lub częściowa kopia chromosomu 21 jest przymocowana do innego chromosomu, najczęściej chromosomu 14. Dziedziczny element został zidentyfikowany w około jednej trzeciej z tych przypadków.
- Mozaiki DS, które występują w około 1% przypadków DS, jest wynikiem niektórych komórek zawierających 46 chromosomów i innych 47.
TS jest zaburzeniem genetycznym dotykającym tylko kobiety. Wynika z brakującego lub częściowo brakującego chromosomu X z większości lub wszystkich komórek. Kobiety z TS nie dojrzewają seksualnie.
Charakterystyka
Fizycznie DS charakteryzuje się niskim napięciem mięśni, lekko spłaszczonym profilem twarzy, w górę oczu, małym wzrostem i pojedynczymi głębokimi zagięciami po środku dłoni. Stan jest związany z opóźnieniami wzrostu fizycznego i łagodnym do umiarkowanej niepełnosprawności intelektualnej, przy czym średnia IQ młodego dorosłego jest odpowiednikiem ośmiu lub dziewięcioletniego (André Mégarbané i in. 2013).
TS charakteryzuje się paszoną szyją, szeroką klatką piersiową, krótkim wzrostem, małym podbródkiem, nieprawidłowymi kątami przedramienia, krótkimi palcami i brakiem dojrzewania seksualnie (Benjamin Saddock i Saddock;. S. Oliveira i Alves).
Diagnoza
Prenatalnie DS można zdiagnozować za pomocą testów surowicy krwi i sonogramu, aby sprawdzić markery. Bardziej inwazyjne i dokładne testy diagnostyczne obejmują przewlekłe pobieranie próbek kosmków, przeprowadzone w pierwszym trymestrze ciąży i amniopunkcję, przeprowadzone w drugim trymestrze trymestru. Oba testy diagnostyczne obejmują 1% ryzyko spontanicznej aborcji, ale oba są prawie w 100% dokładne diagnostycznie. Gdy badanie wskazuje na obecność DS, zarodek jest często przerywany (j. L. Natroli i in.). Po urodzeniu DS identyfikuje się poprzez obecność cech fizycznych i czerpanie krwi z niemowlęcia do testów genetycznych w celu potwierdzenia diagnozy.
Diagnoza TS może wystąpić przy urodzeniu przy użyciu cech fizycznych i pobierania krwi z niemowlęcia do badań genetycznych. Ponieważ większość osób z TS ma normalną inteligencję, warunek można zdiagnozować tylko w okresie dojrzewania z niepowodzeniem początkowym miesiączki; Jednak zauważono, że problemy z wizualizacją przestrzenną są powyżej normy (v.P. Sybert).
Rokowanie
Ponieważ DS i TS są warunkami genetycznymi, nie ma lekarstwa na żadne warunki. Większość ludzi z DS wymaga wsparcia edukacyjnego i chronionego środowiska pracy, aw rozwiniętym świecie ich oczekiwana długość życia wynosiłaby od 50 do 60 lat. W odniesieniu do TS, podczas gdy dziewczęta mogą cieszyć się normalnym życiem z hormonalnymi zabiegami, które pomagają w rozwoju bioder i piersi, nie mogą naturalnie zajść w ciążę.
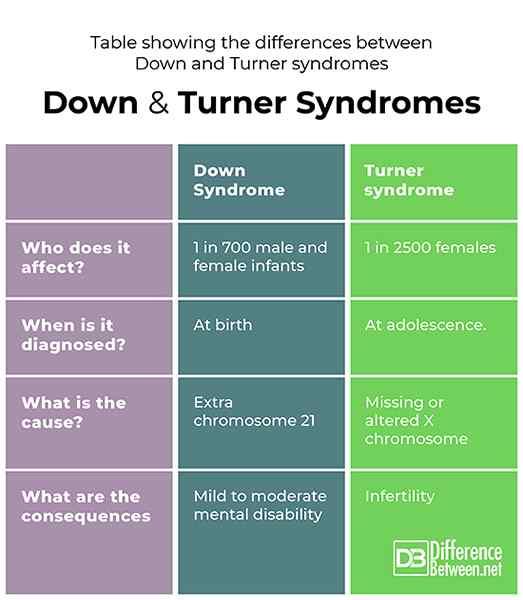
Tabela pokazująca różnice między zespołami w dół i Turnera
Streszczenie
Zespoły Turnera i Downa są spowodowane różnymi nieprawidłowościami chromosomowymi. DS jest wynikiem dodatkowego pełnego lub częściowego chromosomu 21, podczas gdy TS jest wynikiem brakującego lub zmienionego chromosomu X. Na oba płcie mogą mieć wpływ DS, ale TS mogą mieć wpływ tylko kobiety, które mogą zostać zdiagnozowane tylko w okresie dojrzewania. Oba wytwarzają wyraźne cechy fizyczne i chociaż DS jest związane z łagodną i umiarkowaną niepełnosprawnością psychiczną, TS jest związany z niepłodnością.
FAQ
To zespół Turnera i zespół Downa sam?
Zespoły Turnera i Downa należą do najczęstszych nieprawidłowości chromosomalnych zajmowanych przez lekarzy, ale są spowodowane różnymi nieprawidłowościami chromosomowymi. W TS, samice niemowlęcia brakuje chromosomu X, aw DS niemowlę albo płci ma dodatkowy pełny lub częściowy chromosom 21. Chociaż DS jest najczęściej diagnozowane po urodzeniu, zespół Turnera może nie zostać zdiagnozowany do czasu dojrzewania.
Jaka jest różnica między zespołem Downa a zespołem Downa?
Zespół Downa i zespół Downa są synonimami, ale preferencją są zespół Downa, ponieważ stosowanie apostrofu implikuje własność lub posiadanie (National Down Syndrome Society).
Co jest w dół i zespół Turnera?
Zarówno zespoły w dół, jak i Turnera są nieprawidłowościami chromosomowymi, ale chociaż DS jest wynikiem dodatkowego częściowego lub pełnego chromosomu 21, TS jest wynikiem brakującego lub zmienionego chromosomu X w wszystkich lub niektórych komórkach.
Jak zespół Downa różni się od zespołu Klinefelter i Turnera?
Zespół Downa obejmuje dodatkową pełną lub częściową kopię chromosomu 21. Zespół Turnera wpływa tylko na kobiety i obejmuje brakujący lub zmieniony chromosom x we wszystkich lub niektórych komórkach. Zespół Klinefeltera dotyka tylko mężczyzn i obejmuje zarodek otrzymujący dodatkowy chromosom X oprócz chromosomu Y (Benjamin Saddock i Saddock).